Chain Growth Condensation Polymerization
Polymerization can be classified into chain-growth polymerization and step-growth polymerization. Chain growth polymerization is initiated by the reaction of monomer and initiator and then monomers are successively added to the formed radical center to continuously propagate growth. If chain growth proceeds in a living manner, both the molecular weight and composition of the polymer is controlled by the feed ratio of monomers and monomers to initiator. Furthermore, the molecular weight increases directly with monomer conversion while retaining low polydispersity over the whole conversion range. On the other hand, most condensation polymerizations occur via a step-growth mechanism which is a stepwise reaction between bi-functional or multi-functional monomers in which high molecular weight polymers are formed after a large number of steps. Since propagation involves the reaction of all kinds of oligomers with themselves and with monomers, the molecular weight and composition of the polymers is difficult to control, and the polymers possess a broad molecular weight distribution. Thus, a condensation polymerization that occurs by a chain-growth mechanism is highly desirable.
One of the first living chain growth polycondensation methods (CGC) were developed by Yokozawa and Yokoyama.1-3 They proposed two approaches to achieve chain-growth polycondensation of monomers with a nucleophilic and an electrophilic site in each monomer (A-B type):4
Activation of initiator and chain growth center towards reaction with monomer and deactivation of monomer - monomer condensation reactions.
Phase-transfer polymerization (PTP) in two phase system composed of monomer-rich phase and monomer depleted polymerization phase.
In case of method 1, the reaction between AB monomers is suppressed because the B group of the monomer is deactivated by the substituent effect of the A group of the monomer.
Chain Growth Condensation Mechanism
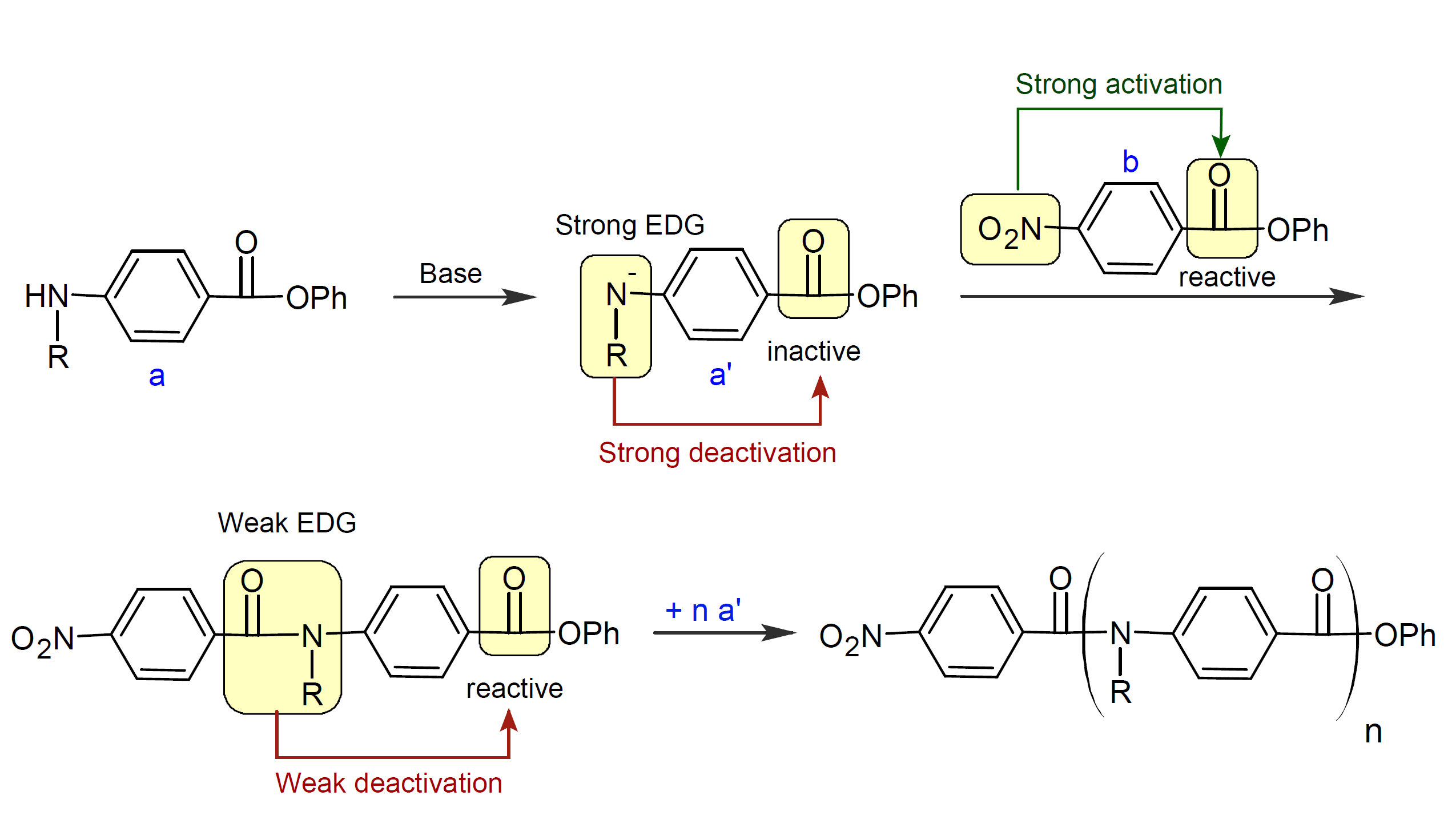
The proposed mechanism of the CGC method is shown in the scheme above for the synthesis of poly(N-alkyl benzamide).1,2 The monomer a is deactivated with a (strong) base such as lithium hexamethyldisilazide which transfers an electron to the amide group of the monomer.5 The resulting amine anion of a' is a strong electron donating group which deactivates the ester group and thus suppresses any reactions between monomers. The ester group of the initiator b, on the other hand, is activated via an electron withdrawing group. Its reactivity is then transferred to the incoming monomers at the chain end which forces the reaction to proceed in a chain growth manner.6
Most of the polymers synthesized in the early studies on CGC contained a long alkyl group on the nitrogen such as octyl.1,2 The long alkyl chain made the polymers more soluble and resulted in higher molecular weights and narrow molecular weight distributions. However, the polymers with pendant long alkyl groups did not have the desired mechanical properties. Therefore, other polymer architectures and functionalities were investigated, such as aramids having more hydrophilic triethylene glycol (TEG) sidechains.7 Other approaches were based on functionalizing the aromatic portion of the aramid. One of the more successful polymers that have been synthesized using the substituent CGC technique, is polybenzoxazole.8 Other classes of polymers produced via substituent effect-assisted chain-growth condensation polymerization include aromatic polyesters,2 polyethers,9 polyphenylene10 and polyethersulfones11. The CGC method has also been employed to synthesize well-defined block copolymers and star-shaped polymers.12
In case of phase transfer polymerization, monomers are “confined” in a solid or liquid
phase, in which the monomers do not react with each other. The
second phase is a monomer depleted liquid to which an initiator is
added. Polymerization starts when monomers migrate through the phase
boundary and react with the initiator. If the polymer end groups are
more reactive than the monomers, and the rate of diffusion through
the boundary is adequate, monomers predominantly react with the
polymer end groups, resulting in polymers with low polydispersity. This method is not new, but as Yokozawa and Yokoyama2 pointed out, traditional
phase transfer polymerization13,14, has given little to no control over molecular weight and polydispersity of condensation polymers, whereas good control over MW has been achieve when utilizing the substituent effect.
Notes & References:
T. Yokozawa, T. Asai, R. Sugi, S. Ishigooka, and S. Hiraoka, J. Am. Chem. Soc., 122, 8313-8314 (2000)
T. Yokozawa and A. Yokoyama, Poly. J., Vol. 36 (2), 65—83 (2004)
A. Yokoyama, T. Yokozawa, Macromolecules, 40, 12, 4093-4101 (2007)
The observation that end groups of condensing polymers are more reactive than monomers had been reported by several other scientist earlier. However, none of their methods yielded polymers with precisely controlled molecular weight and low polydispersity due to the insolubility of the growing polymers and due to contaminations.
T. Yokozawa, D. Muroya, R. Sugi, A. Yokoyama, Macromol. Rap. Comm., Vol. 26 (12), 976-981 (2005)
The rate of polymerization strongly depends on the type of side chain. For example, monomers with a tri(ethylene glycol) monomethyl ether (TEG) side chain instead of an alkyl side chain polymerize much slower.7
J.R. Ruiz, M. Trigo-Lopez, F.C. Garcia, J.M. Garcia, Polymers, 9, 414 (2017)
Y. Ohta, T. Niiyama, A. Yokoyama and T. Yokozawa, J. of Poly. Sci. Part A: Poly. Chem., 2014, 52, 1730-1736
T. Yokozawa, Y. Suzuki, and S. Hiraoka, J. Am. Chem. Soc., 123, 9902 (2001)
R. Miyakoshi, K. Shimono, A. Yokoyama, T. Yokozawa, J. Am. Chem. Soc., 128, 50, 16012-16013 (2006)
A. Benhalima , A. Furtos , J. Brisson, Macromol. Chem. Phys., 214, 2489-2499 (2013)
A. Yokoyama, and T. Yokozawa, Macromolecules, 40, 12, 4093-4101 (2007)
V. Percec, T.D. Shaffer, and H. Nava, J. Polym. Sci., Part C, Polym. Lett., 22 (12), 637-647 (1984)
T.D. Shaffer and M.C. Kramer, Makromol. Chem., 191 (1), 71-79 (1990)
March 28, 2020